 基因分型方法.ppt
基因分型方法.ppt
《基因分型方法.ppt》由会员分享,可在线阅读,更多相关《基因分型方法.ppt(82页珍藏版)》请在第壹文秘上搜索。
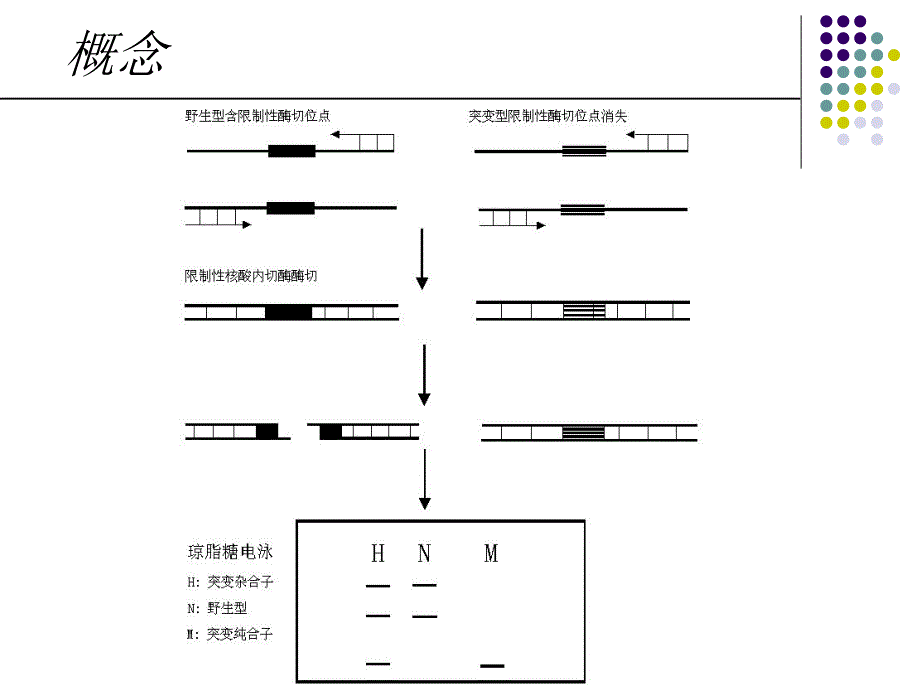
1、PCR-RFLP 基因分型方法基因分型方法-关于序列查找、引物设计和酶切位点分析概念聚合酶链式反应连接的限制性片段长度多态性分析 限制性片段长度多态性(restrition fragment length polymophism,RFLP):是指由限制性酶切位点间的插入缺失重排或点突变所引起的基因型间限制性片段长度的变异。PCR-RFLP:是在 PCR 和 DNA 序列分析基础上产生的 RFLP 技术。该方法是通过 PCR 扩增一段 DNA 片段,然后再选择适当的限制性内切酶,消化 PCR 产物,经电泳,可得到有特异性的电泳谱带,从而达到鉴定不同基因型的目的。概念概念确定基因序列设计引物选择内
2、切酶电 泳基 因 型Step by step基因序列查找Exon 1Exon 2Exon 3PromotergDNAcDNAPromoter基因序列查找http:/www.genomatix.de/http:/www.ncbi.nlm.nih.gov/Genbankhttp:/expasy.org/sprot/基因序列查找选择gene输入基因名称基因序列查找基因的全称基因的物种来源基因序列查找cDNA序列gDNA序列基因序列查找序列如何详细区分每一个外显子和内含子?基因序列查找基因序列查找外显子全序列基因序列查找包含有该外显子的序列该外显子的全序列基因序列查找起始密码子外显子内含子启动子?启动
3、子序列查找先免费注册登录启动子序列查找启动子序列查找选择Gene2Promoter启动子序列查找选择物种基因的名字启动子序列查找提交选择你的基因继续启动子序列查找启动子序列查找可以选择是否分析该启动子不同颜色代表该启动子序列的可靠性,金黄色代表实验研究证明启动子序列查找开始分析启动子序列查找继续提交点击获得序列启动子序列查找启动子序列查找详细序列如何确定我的SNP位置C218A:外显子编码方式rs431898:pubmed SNPs数据库编号 C-256A:启动子编码方式如何确定我的SNP位置ATG的A是第一位C218AC-256A如何确定我的SNP位置选择SNP选项输入编号rs.如何确定我的
4、SNP位置该SNP附近的序列关于该SNPs的详细信息如何预知我的SNP的功能意义输入蛋白的名称如何预知我的SNP的功能意义如何预知我的SNP的功能意义如何预知我的SNP的功能意义结构域名称起止氨基酸编号该结构域的氨基酸长度如何预知我的SNP的功能意义多态性包括SNPs定点诱变研究氨基酸的改变是否有功能意义基因序列查找更多序列查找方法、查找途径、数据库信息和序列比对请参考引物设计引物设计引物一般原则基本原则:1.引物要跟模板紧密结合,2.引物与引物之间不能有稳定的二聚体或发夹结构存在,3.引物不能在别的非目的位点引起DNA聚合反应(即错配)。需要考虑的因素:引物长度(primer length)
5、、产物长度(product length)、序列Tm值(melting temperature)、G值(internal stability)、引物二聚体及发夹结构(duplex formation and hairpin)错误引发位点(false priming site)、引物及产物GC 含量(composition)引物一般原则引物的长度:15-30bp,常用的是18-27bp 太短:特异性不好 太长:延伸温度大于Taq酶的最适温度 引物末端要求:3端的序列要比5端重要,引物3端的碱基一般不用A A在错误引发位点的引发效率相对比较高,5端序列对PCR 影响不大,常用来引进修饰位点或标物。
6、引物的GC含量:一般为40-60%,以45-55为宜 两条引物之间的GC含量不宜相差太大 引物一般原则Tm 值:尽量保证两条引物的Tm值相匹配。最 好相差不要大于5 度引物二聚体及发夹结构的能量:绝对值一般不要超过4.5 最好不要位于3末端否则容易产生引物二聚体带而且会降低引物浓度从而导致PCR 正常反应不能进行,如果在5末端对反应就没有多大的影响了 G值:反映了引物与模板结合的强弱程度。一般情况下,引物的 G值最好呈正弦曲线形状,即5端和中间G值较高而 3端G值相对较低,且不要超过9。如此则有利于正确引 发反应而可防止错误引发 PCR-RFLP 引物特殊要求PCR产物的长度:200-1000




- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 基因 方法
 第壹文秘所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
第壹文秘所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 重点工作绩效评估自评表.docx
重点工作绩效评估自评表.docx
-
2022 WHO成人、青少、儿童HIV感染者隐球菌病的诊断、预防和管理(全文).docx
-
国家电网行测专项练习题二.docx
-
铁路安全保证书范文.docx
-
2022 ESC室性心律失常管理和心脏性猝死预防指南要点(全文).docx
-
宁夏省安全员C类考试试题.docx
-
2022 EASL消除病毒性肝炎总结(全文).docx
-
2021煤炭工业露天矿建设项目考试答案.docx
-
2022 BSG功能性消化不良的管理要点(全文).docx
-
2022中国原发性结直肠癌规范诊疗质量控制指标(全文).docx
-
2022不同移植胚胎类型和数量与异位妊娠发生风险相关性的研究进展(全文).docx
-
2022YIIN道分娩宫缩乏力性产后出血的早期识别及处理(全文).docx
-
2022中国膀胱癌保膀胱治疗多学科诊治协作共识(全文).docx
-
2022中国宫颈癌规范诊疗质量控制指标(最全版).docx
-
2022中国噬血细胞综合征诊断与治疗指南(最全版).docx
-
2022某县纪委监委关于漠视侵害群众利益问题专项整治工作情况汇报.docx
-
2022某市某区统筹疫情防控和稳定经济增长社会发展工作实施方案2篇.docx
-
2022某市关于贯彻落实全国稳住经济大盘电视电话会议精神和全省稳经济工作电视电话会议贯彻落实情况的报告.docx
-
2022民生实事项目任务分解方案.docx
-
2022某局以案促改工作开展情况报告(含主要做法下一步计划及打算).docx
-
2022正确认识和把握我国发展重大理论和实践问题学习心得感悟范文.docx
-
2022检察机关扫黑除恶专项斗争工作开展情况的报告.docx
-
2022查摆理论学习思想状态组织纪律三个方面问题专题组织生活会个人对照检查材料.docx
-
2022法治政府建设工作总结.docx
-
2022校长述职报告范文.docx
-
2022母婴安全保障自查自纠报告(工作开展情况、存在问题及下步整改措施计划).docx
-
2022机关党建工作要点三篇(1).docx
-
2022某县某市“六稳”“六保”工作开展落实情况汇报材料2篇.docx
-
“两在两同”我先行—区委统战部“同心筑梦”行动实施方案.docx
-
“三重一大”决策制度整改汇报.docx
-
“两个确立”专题党课讲稿6篇汇编.docx
-
“三强化”抓实党风廉政建设.docx
 中班语言诗歌《伞》PPT课件教案PPT课件.pptx
中班语言诗歌《伞》PPT课件教案PPT课件.pptx
-
中班语言活动说课稿《萝卜回来了》PPT课件教案幼儿教育课件萝卜回来了.pptx
-
中班语言绕口令《高高山上一条藤》PPT课件教案音效动画PPT.pptx
-
中班语言诗歌《爸爸妈妈和我》PPT课件音乐PPT课件.pptx
-
中班语言艺术《漂亮的蝴蝶结》PPT课件教案漂亮的蝴蝶结[1].pptx
-
中班语言说课《大卫不可以》PPT课件大卫不可以-说课.pptx
-
中班语言课件《春雨的吉他》PPT课件教案中班语言《春雨的吉他》课件.pptx
-
中班语言诗歌《天的画报》PPT课件教案中班综合:天的画报.pptx
-
中班语言课件《小兔子开铺子》PPT课件教案中班语言《小兔子开铺子》课件.pptx
-
中班语言诗歌《伞》PPT课件中班诗歌《伞》.pptx
-
中班语言诗歌《谁的耳朵》PPT课件教案音乐谁的耳朵.pptx
-
中班语言诗歌《小小鸟捡花瓣》PPT课件教案小小鸟捡花瓣.pptx
-
中班语言诗歌《家》PPT课件教案配音音乐PPT课件.pptx
-
中班语言课件《如果我能飞》PPT课件教案中班语言《如果我能飞》课件.pptx
-
中班语言诗歌课件《家》PPT课件教案中班语言诗歌《家》课件.pptx
-
中班语言说课稿《亲爱的护士小姐》PPT课件教案亲爱的护士小姐说课稿.pptx
-
中班语言诗歌《家》PPT课件教案音乐PPT课件.pptx
-
中班语言说课稿《萝卜回来了》PPT课件教案中班语言萝卜回来了说课课件.pptx
-
中班语言课件《谁的本领大》PPT课件教案中班语言《谁的本领大》课件.pptx
